Genomics
基因组学 (Genomics) 是对一个人的所有基因(即基因组)的研究,包括这些基因之间以及与基因与外界环境之间的相互作用。
——美国国立卫生研究院(NIH)
基因组 Genome

1952年,Rosalind Franklin获得了B型DNA X射线晶体衍射照片,确认了DNA的螺旋结构;1953年,James D. Watson 和Francis Crick发表了DNA结构;1955年,Fred Sanger发表了胰岛素氨基酸序列。自此,核酸测序成为早期分子生物学家的主要目标。基因组是生物体的完整DNA集,包括其所有基因;基因组学的目标是对生物体的所有基因、及其相互关系和对生物体的影响进行定性和量化。



测序技术的发展
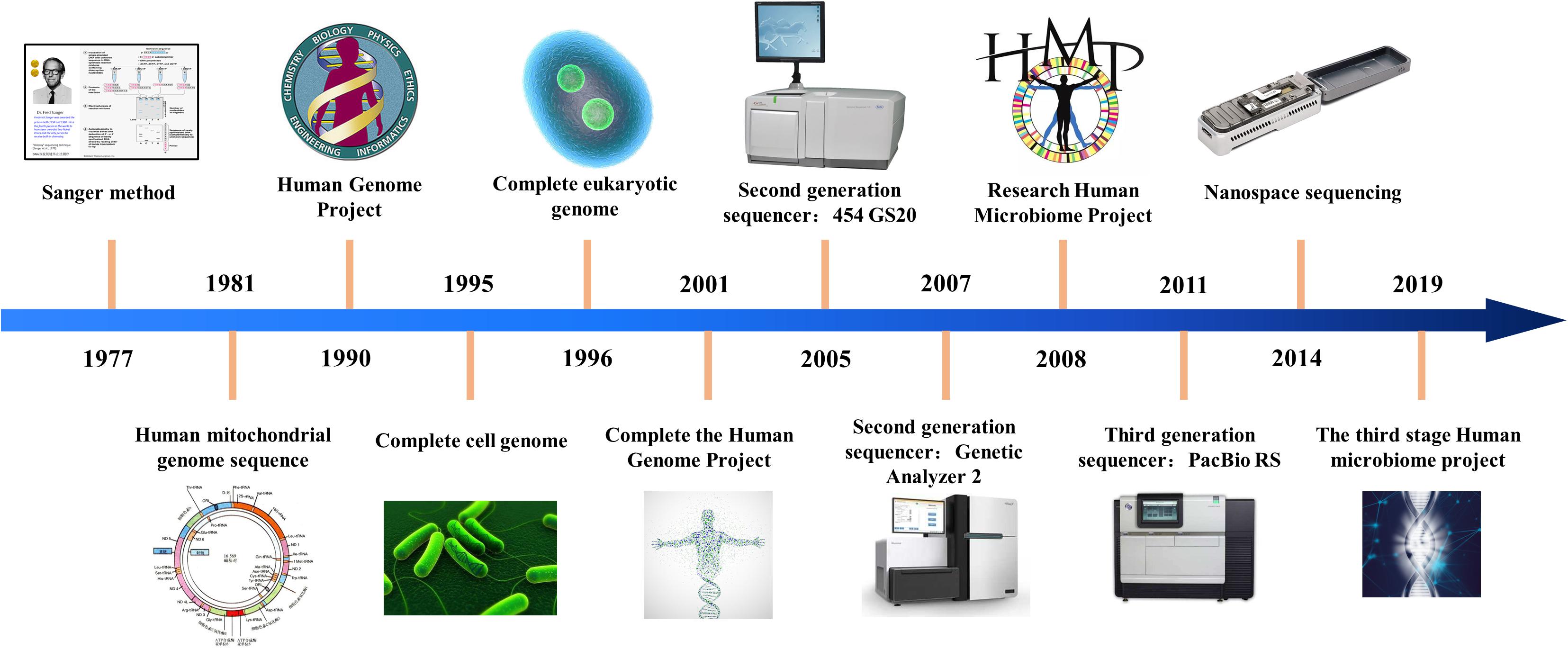
序列的测定是基因组学研究的最基础也最重要的工作。1977年Sanger提出了“链终止法”,这种测序方法被称为第一代测序技术,该技术直到现在依然被广泛使用,但是一次只能获得一条长度在700~1000个碱基的序列,无法满足现代科学发展对生物基因序列获取的迫切需求。
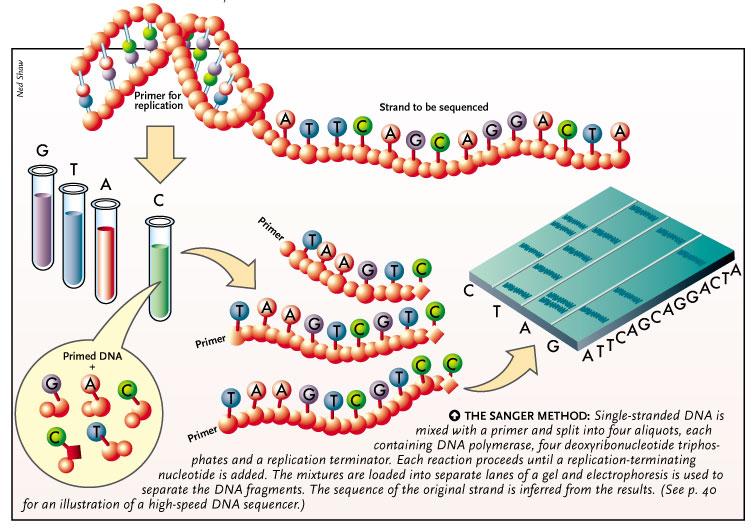
双脱氧链终止法:采用DNA复制原理,测序反应体系中包括目标DNA片段、脱氧三磷酸核苷酸(dNTP)、双脱氧三磷酸核苷酸(ddNTP)、测序引物及DNA聚合酶等。其技术核心是:ddNTP缺少3'-OH基团,不具有与另一个dNTP连接形成磷酸二酯键的能力,这些ddNTP可用来中止DNA链的延伸;同时,这些ddNTP上连接有放射性同位素或荧光标记基团,因此可以被自动化的仪器或凝胶成像系统所检测到。
高通量测序 (High-Throughput Sequencing, HTS)是对传统Sanger测序的革命性变革,其解决了一代测序一次只能测定一条序列的限制,一次运行即可同时得到几十万到几百万条核酸分子的序列,因此也被称为新一代测序 (Next Generation Sequencing, NGS)或第二代测序。第二代测序技术虽然测序的通量大大增加,但是其获得单条序列长度很短,想要得到准确的基因序列信息依赖于较高的测序覆盖度和准确的序列拼接技术,因此最终得到的结果中会存在一定的错误信息。
第三代测序技术也称为单分子测序技术,该技术在保证测序通量的基础上,对单条长序列进行从头测序,能够直接得到长度在数万个碱基的核酸序列信息。
全基因组测序与组装
全基因组测序主要包括样本获取、细胞裂解(DNA提取)、全基因组扩增与核酸纯化(测序文库制备)及序列测定四个部分。测序样本是的获取是基因组学研究的第一步,也是关系到基因组质量的最重要的一步。可以根据需求选取某一群体作为样本(宏基因组学, Metagenomics)或者以特定细胞为样本( 单细胞基因组学, Single-cell genomics )。宏基因组学样本可依据细胞丰度选择是否进行全基因组扩增,如果细胞丰度大可跳过全基因组扩增直接将细胞裂解后提取的DNA作为测序文库;而单细胞由于核酸量微少需要进行全基因组扩增。目前市场上已经有成熟的试剂盒可用于全基因组测序文库的制备。然而,不论是宏基因组还是单细胞基因组分析,都应尽量避免污染,以免对基因组质量造成影响。
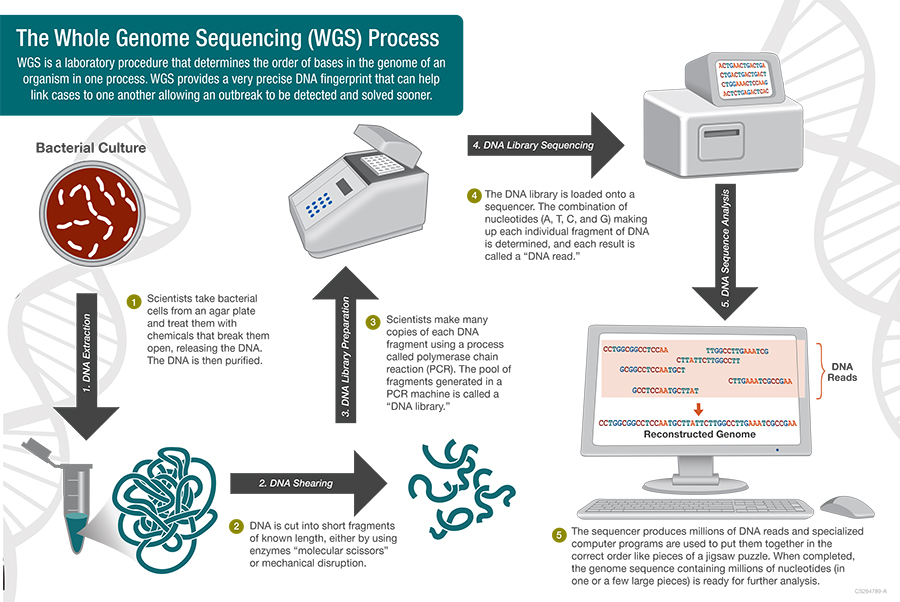
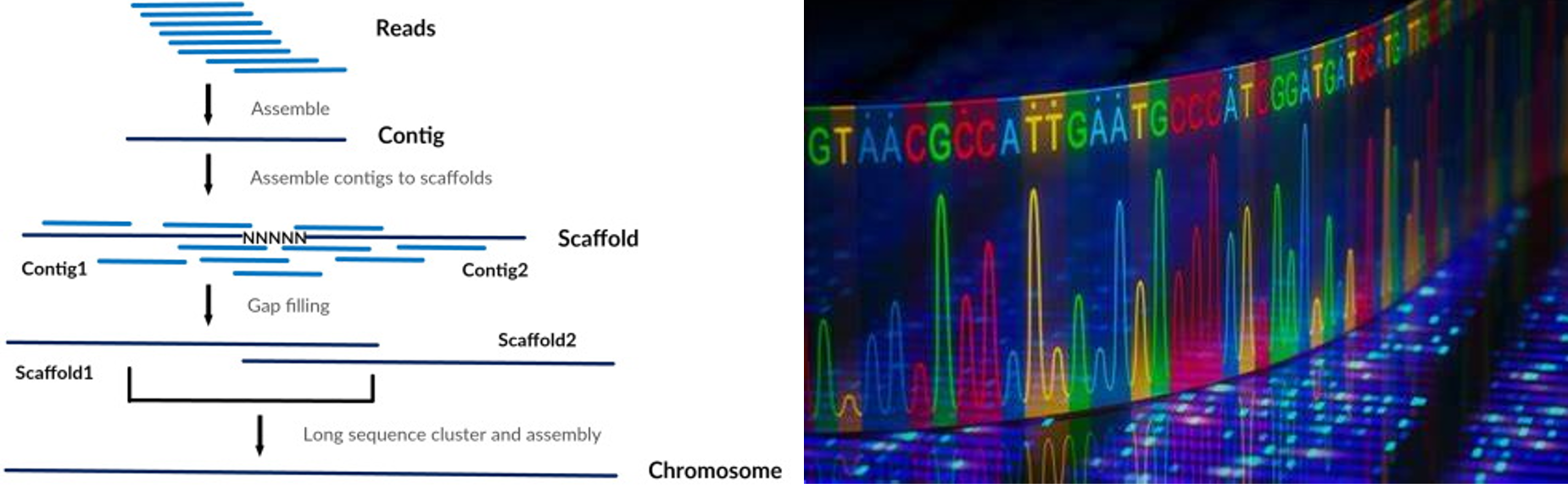
尽管目前第三代测序技术如PacBio或Oxford Nanopore能够获得超过10kb的序列,但是测序结果往往存在较高的错误率,约15%(详见),因此,目前第一、第二代测序技术仍然是主流的测序手段。测序完成后,需要对得到的短序列核酸片段(reads)进行组装从而获得基因组草图。基因组的从头组装(de novo assembly)过程是对reads进行组装产生长片段Contigs,然后确定Contig的方向和顺序,组装Contigs产生更长的片段Scaffolds,最后再组装连接Scaffolds得到完整的染色体序列。最常用的从头组装算法是Overlap-Layout-Consensus(OLC)算法,依据序列片段的重叠区(overlap)进行拼接,逐渐得到基因组草图。
基因组学研究方向
目前基因组学的研究方向包括功能基因组学、比较基因组学等,研究成果可用于疾病治疗、药物开发、生物工程及基因组编辑与修饰等领域。